La fibrosi cistica è la malattia genetica grave più diffusa.
E’ una patologia multiorgano, che colpisce soprattutto l’apparato respiratorio e quello digerente. E’ dovuta ad un gene alterato, cioè mutato, chiamato gene CFTR (Cystic Fibrosis Transmembrane Regulator), che determina la produzione di muco eccessivamente denso. Questo muco chiude i bronchi e porta a infezioni respiratorie ripetute, ostruisce il pancreas e impedisce che gli enzimi pancreatici raggiungano l’intestino, di conseguenza i cibi non possono essere digeriti e assimilati.
Seppure il grado di coinvolgimento differisca anche notevolmente da persona a persona, la persistenza dell’infezione e dell’infiammazione polmonare, che causa il deterioramento progressivo del tessuto polmonare, è la maggior causa di morbilità nei pazienti FC. Le manifestazioni tipiche della malattia sono:
- difficoltà nella digestione dei grassi, proteine, amidi
- carenza di vitamine liposolubili
- perdita progressiva della funzione polmonare
La malattia non danneggia in alcun modo le capacità intellettive e non si manifesta sull’aspetto fisico né alla nascita né in seguito nel corso della vita, per questo viene definita la “malattia invisibile”.
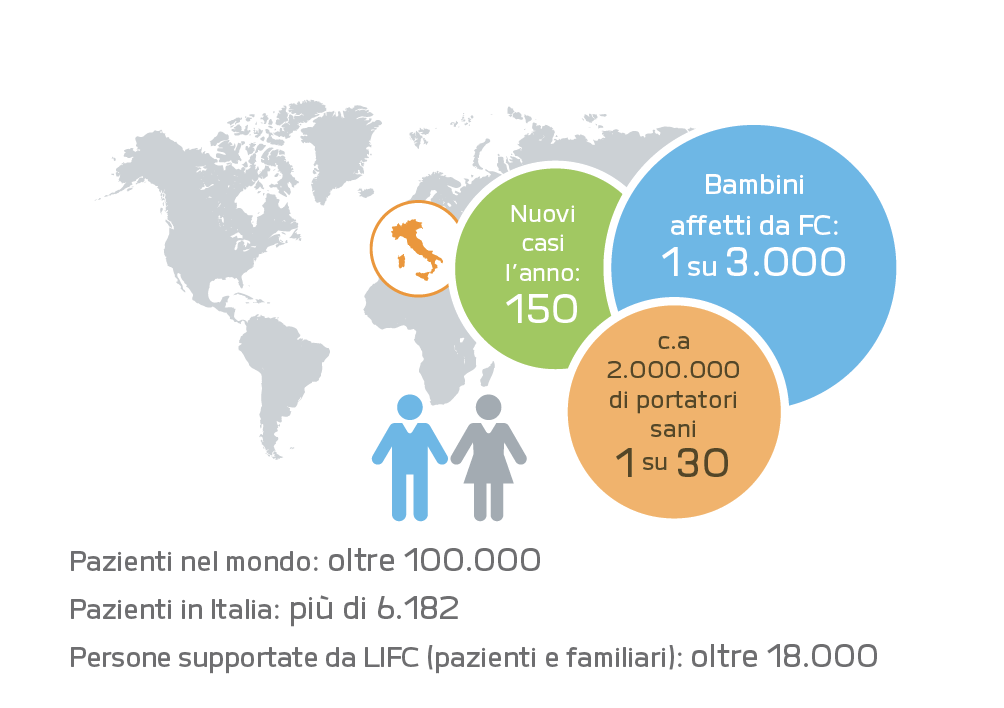
DIFFUSIONE DELLA MALATTIA
DIFFUSIONE DELLA MALATTIA
Si stima che ogni 2.500-3.000 bambini nati in Italia, 1 è affetto da fibrosi cistica (circa150 nuovi casi all’anno).
La malattia colpisce indifferentemente maschi e femmine. Oggi quasi 6.000 bambini, adolescenti e adulti affetti da FC vengono curati nei Centri Specializzati in Italia.
Per merito dei continui progressi terapeutici, assistenziali, nonché diagnostici, la prevalenza dei pazienti FC in Italia, con età uguale e superiore ai 18 anni, è del 65,2% (Report at-a-glance RIFC anno 2024)
CAUSE DELLA FIBROSI CISTICA
La malattia si manifesta quando un bambino eredita due copie alterate, cioè mutate, del gene CFTR, una da ciascun genitore. Il gene CFTR codifica la sintesi della proteina CFTR, che se ben funzionante, regola il movimento del cloro, al quale segue il movimento dell’acqua, dall’interno verso l’esterno delle cellule epiteliali delle ghiandole mucose.
I genitori che hanno solamente una copia alterata del gene CFTR non hanno la fibrosi cistica, né evidenziano alcun sintomo della malattia e sono definiti portatori sani del gene della fibrosi cistica.Possono però trasmettere il gene difettoso ai figli, così come trasmettono altre caratteristiche, come il colore degli occhi e dei capelli.
La frequenza dei portatori sani di mutazioni del gene FC in Italia e nel mondo occidentale è approssimativamente di 1 ogni 30 persone.Quando due genitori portatori sani, cioè portatori entrambi di una mutazione, hanno un figlio, esiste 1 probabilità su 4 che il bambino nasca con FC.
I sintomi della fibrosi cistica
Il malfunzionamento o l’assenza della proteina CFTR interessa tutte le ghiandole a secrezione mucosa determinando una carenza di cloro e acqua nelle secrezioni. Le secrezioni mucose povere di acqua e vischiose tendono a ristagnare provocando così l’ostruzione degli organi interessati, prevalentemente bronchi, intestino e pancreas. Nell’albero bronchiale (il cui grado di coinvolgimento è il principale responsabile della gravità della malattia), si innesca un circolo vizioso infiammazione-infezione che porta infine alla destrutturazione del tessuto polmonare. La fibrosi cistica è una malattia che colpisce molti organi e che produce una varietà di sintomi tra cui:<
- Tosse persistente dapprima stizzosa poi catarrale
- Respiro sibilante e affanno
- Infezioni bronchiali e polmonari frequenti
- Diarrea cronica con emissione feci oleose e maleodoranti, in alcuni casi ostruzione intestinale alla nascita (ileo da meconio) e spesso ostruzioni intestinali ripetute in età adolescenziale e adulta
- Scarso accrescimento in peso e altezza
- Sudore salato
Esiste una cura per la fibrosi cistica?
La terapia di questa malattia ha avuto negli ultimi anni un notevole sviluppo.
Infatti, accanto ad una terapia dei sintomi adesso si comincia a disporre di terapie personalizzate che curano il difetto di base in alcune forme geniche di FC e si spera che, entro alcuni anni, tutte le mutazioni genetiche saranno curabili.
Contenuti realizzati a cura della Prof.ssa Serena Quattrucci, Consulente Scientifico LIFC, ultimo aggiornamento Giugno 2023.